


A fibrodisplasia ossificante progressiva é uma doença genética rara do tecido conjuntivo, caracterizada por ossificação disseminada em tecidos moles e alterações congênitas das extremidades. Sua transmissão é autossômica dominante, com penetrância completa, mas expressão variável.
A expressão fibrodisplasia ossificante progressiva substitui a antigo nome miosite ossificante, por ser a osteogênese ectópica ocorrendo no tecido conjuntivo, podendo afetar músculos, fáscias, ligamentos, tendões e cápsulas articulares.
O doente geralmente apresenta ossificação progressiva do tecido conjuntivo, que causa uma limitação crescente da mobilidade osteoarticular, afetando principalmente a coluna vertebral, ombros, quadril e articulações periféricas.
O paciente assume uma postura única e limitada, não podendo sequer sentar. Este aspecto caracteriza a forma avançada da doença, recebendo a denominação de síndrome do “stone man”, antes descrita em apenas cerca de 600 doentes. Nesse estágio, o paciente rapidamente evolui para a morte, em conseqüência de problemas respiratórios de origem restritiva.
 |
2.1. O QUE É FIBRODISPLASIA OSSIFICANTE PROGRESSIVA?
Fibrodisplasia Ossificante Progressiva (FOP) é uma doença genética rara que causa a formação de ossos no interior dos músculos, tendões, ligamentos e outros tecidos conectivos. Pontes de ossos "extra" se desenvolvem através das articulações (juntas do corpo) restringindo progressivamente os movimentos.
Fibrodisplasia Ossificante Progressiva (FOP) é uma doença genética rara que causa a formação de ossos no interior dos músculos, tendões, ligamentos e outros tecidos conectivos. Pontes de ossos "extra" se desenvolvem através das articulações (juntas do corpo) restringindo progressivamente os movimentos.
Na FOP, o corpo não somente produz muitos ossos, mas um todo um esqueleto "extra" é formado, envolvendo o corpo e prendendo a pessoa em uma prisão de ossos para a qual não existe uma chave.
A fibrodisplasia ossificante progressiva (FOP) é uma doença rara, autossômica dominante, com expressão variável, que afeta todos os grupos étnicos. Sua prevalência é de 0,61 caso por um milhão de habitantes.
Como na maioria dos casos não se observam antecedentes familiares, imagina-se ser esta doença devida a uma mutação esporádica. É uma enfermidade incapacitante em crianças e adultos jovens, basicamente caracterizada por osteogênese heterotópica progressiva e alteração congênita dos primeiros pododáctilos.
2.2. DESCOBRINDO O CÓDIGO GENÉTICO
A pesquisa sobre a FOP é um trabalho de detetive. O principal objetivo é encontrar o caminho certo através do labirinto genético para identificar o gene danificado que causa a produção aumentada de uma poderosa proteína produtora de ossos nas pessoas com FOP.
Como em outras doenças genéticas, a pesquisa da FOP envolve o encontro e conserto de uma chave genética defeituosa.
Ao mesmo tempo, entretanto, a FOP é também uma doença única. Não existe nenhuma outra condição em que um tipo de tecido do corpo se transforma em outro da mesma forma que no caso da FOP, os músculos se transformam em ossos. Apesar disso, a equipe de pesquisa tem feito avanços significativos em mapear o possível gene da FOP e isto traz uma enorme esperança à comunidade FOP.
As crianças com FOP parecem normais ao nascimento, exceto por uma malformação congênita dos dedos grandes dos pés. Durante a primeira ou segunda década de vida, nódulos fibrosos dolorosos se desenvolvem pelo pescoço, costas e ombros. Estes nódulos dão lugar a ossos, em um processo conhecido como ossificação heterotópica. A FOP então progride pelo tronco e membros do corpo, substituindo os músculos saudáveis por ossos de aparência normal.
As crianças com FOP parecem normais ao nascimento, exceto por uma malformação congênita dos dedos grandes dos pés. Durante a primeira ou segunda década de vida, nódulos fibrosos dolorosos se desenvolvem pelo pescoço, costas e ombros. Estes nódulos dão lugar a ossos, em um processo conhecido como ossificação heterotópica. A FOP então progride pelo tronco e membros do corpo, substituindo os músculos saudáveis por ossos de aparência normal.
Estas pontes de ossos restringem significativamente os movimentos, entretanto, qualquer tentativa de remover estes ossos resulta em uma formação explosiva de novos ossos, Isto ocorre por que um trauma, como uma cirurgia (e até mesmo uma batida, queda ou injeções intramusculares) acelera o processo da FOP.
A FOP é extremamente variável e imprevisível. Em algumas pessoas, a progressão da doença é rápida, enquanto em outras é gradual. Algumas pessoas passam meses e até mesmo anos sem apresentar um surto (flare-up), enquanto outras formam ossos extra o tempo todo. Em todos os casos, ossos podem se formar a qualquer momento e em qualquer músculo, com ou sem trauma precedente.
Por todo o mundo, a FOP afeta uma a cada 2 milhões de pessoas.
As pesquisas sobre a FOP estão fazendo descobertas sobre o processo básico de formação óssea.
3. O PRIMEIRO CASO DE FOP
Em 1692, o médico francês Guy Patin foi o primeiro a registrar um caso de FOP. O primeiro caso documentado de maneira completa da doença aconteceu em 1740, quando um médico de Londres descreveu um adolescente com grandes inchaços de ossos no corpo em uma carta para a Universidade Real de Médicos [fonte: IFOPA (em inglês)].
4. CASO CLÍNICO
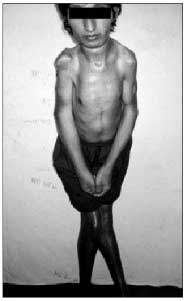
Paciente de 22 anos de idade, branco, primeiro filho de uma prole de três (irmãos normais), sexo masculino, foi atendido em 21 de janeiro de 2002 no ambulatório de Ortopedia do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), referindo dor nas articulações e dificuldade para andar e principalmente ao sentar. Foi aparentemente saudável até os oito anos de idade, quando passou a ter episódios recorrentes de dor, rigidez das pernas e da coluna, evoluindo posteriormente para todo o corpo. O paciente não tinha antecedente pessoal ou familiar relevantes.
4.1. EXAME FÍSICO ESPECÍFICO
• Grande limitação funcional global do paciente, com rigidez universalmente distribuída.
• Ausência de mobilidade do esqueleto axial e redução muito significativa da mobilidade das articulações periféricas.
• A articulação temporomandibular estava limitada e condicionava redução da abertura bucal.
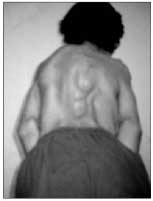
• A coluna vertebral apresentava-se anquilosada, com escoliose torácica à esquerda, acentuação da lordose lombar e focos de ossificações ectópicas na musculatura paravertebral
• Redução da expansibilidade torácica.
• Evidente rigidez nos quadris e ombros.
• Importante atrofia muscular.
• Adução das coxas.
• Genuvalgo bilateral.
• Pés em equinovaro.
 |  |
Radiografia dos pés, incidência antero- posterior.Anomalia congênita característica dos primeiros dedos, com encurtamento da falange proximal do hálux e valgismo (setas)
 |
As radiografias documentaram múltiplas calcificações em partes moles no pescoço.
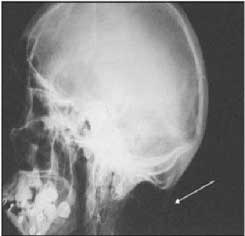 |
Radiografia da coluna cervical, incidência ântero-posterior. Ossificações das costelas e fusão das uncoarticulações.
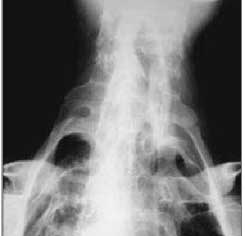 |
Radiografia do fêmur, incidência ântero-posterior. Presença de ossificações cilíndricas em partes moles de ambas as coxas (setas retas), com pseudo-artroses nos ossos do quadril.
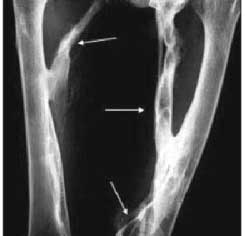 |
Radiografia dos joelhos, incidência ântero- posterior. Ossificações periarticulares com anquilose das articulações.
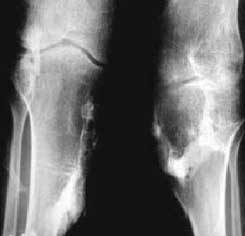 |
4.2. CASO CLÍNICO: MANUEL
 | Manuel vive na Argentina e tinha apenas 4 anos quando lhe foi diagnosticado Fibrodisplasia Ossificante Progressiva (FOP), uma doença genética extremamente rara que afeta 1 pessoa em cada 2 milhões. Manuel teve o seu primeiro episódio doloroso em 2000, quando tinha 3 anos. Estes episódios ocorrem quando o corpo começa a produzir osso novo, o que provoca o edema (inchaço) dos tecidos. |
Manuel teve o seu primeiro episódio doloroso em 2000, quando tinha 3 anos. Estes episódios ocorrem quando o corpo começa a produzir osso novo, o que provoca o edema (inchaço) dos tecidos. Normalmente este processo é bastante doloroso. Inicialmente suspeitou-se que os sintomas que apresentava eram sinais de cancro. «O Manuel foi submetido a bastantes exames», explica a sua mãe, Moira. «Até uma biopsia fez, o que é completamente desaconselhado no caso da FOP, porque o choque da biopsia provoca a formação de osso novo no local do impacto. O primeiro diagnóstico indicava que ele tinha miofibromatose, a que se seguiu outro de fasceíte. Comecei por enviar 800 mensagens de correio eletrônico para hospitais de todo o mundo, com uma descrição dos sintomas que o meu filho apresentava! Nunca ninguém tinha visto nada assim, por isso começávamos a pensar que o diagnóstico inicial estava errado. Uma das respostas que recebemos sugeria o nome de um médico na Argentina e foi ele que finalmente fez o diagnóstico correto em Março de 2001.
A FOP partilha muitas características adversas com outras doenças raras e muito raras: atrasos nos diagnósticos, que por vezes são incorretos, inexistência de tratamento, poucos conhecimentos científicos e falta de informação sobre a doença entre os profissionais de saúde. Pouco depois do diagnóstico, Moira e o seu marido tiveram conhecimento da International Fibrodysplasia Ossificans Progressiva Association - IFOPA (Associação Internacional da Fibrodisplasia Ossificante Progressiva). |  |
A associação deu-nos toda a informação de que precisávamos. Tivemos de aprender muitas coisas novas, tal como adaptar a casa para o Manuel e como falar sobre a FOP com ele e com a nossa família... Tivemos a sorte desta comunidade pró-ativa ter feito importantes avanços nos últimos 16 anos em relação aos conhecimentos sobre a doença», continua Moira. A IFOPA tem membros em 50 países diferentes e o seu sítio web tem fóruns de discussão em que as pessoas podem trocar informações em espanhol, inglês e português. Moira Liljesthröm acabou por ser um membro ativo da comunidade da FOP ao ajudar a criar o grupo latino-americano da FOP (ALAFOP), em 2003, uma rede de cerca de 80 pessoas afetadas pela doença, de 10 países latino-americanos. Mais recentemente, ela e o marido fundaram a Fundación FOP na Argentina, que, para além de se dedicar à FOP, está a levar a cabo um projecto de investigação sobre a situação social, médica e legal das pessoas afetadas pelas doenças raras na Argentina. Este projeto é financiado pela Agência Científica e Tecnológica do Governo Federal da Argentina. No que respeita a este projeto, Moira pensa que a Eurordis pode ajudar a Fundación FOP. «Podíamos aproveitar a experiência da Eurordis relativamente à forma de realizar inquéritos de investigação, como os inquéritos EurordisCare 1 e EurordisCare 2. Penso que a cooperação entre redes, grupos e associações poderá ajudar a colocar as doenças raras nas agendas de saúde pública e a aumentar o grau de conscientização sobre elas.
Uma das razões para continuar a ter esperança diz respeito à descoberta do gene da FOP, em 2006, por investigadores da Faculdade de Medicina da Universidade da Pensilvânia, nos EUA. A descoberta do gene da FOP e da mutação que provoca a FOP proporciona um alvo bastante específico para o desenvolvimento futuro de medicamentos que não se limitem a tratar os sintomas mas que tratem a própria doença. Esta investigação foi parcialmente financiada pela IFOPA. Atualmente, o Manuel está no 5.º ano e freqüenta uma escola com um ambiente seguro para limitar os traumatismos que ele possa sofrer. O Manuel tem alguma dificuldade em levantar os braços e em dobrar o tronco, mas isso não o impede de brincar e de fazer muitas outras coisas! conclui a sua orgulhosa mãe.
5. SINAIS E SINTOMAS
Os sintomas da FOP são variáveis, com a maioria dos doentes apresentando calcificação das partes moles antes mesmo dos dez anos de idade.
5. SINAIS E SINTOMAS
Os sintomas da FOP são variáveis, com a maioria dos doentes apresentando calcificação das partes moles antes mesmo dos dez anos de idade.
A idade média de início é em torno dos 3,6 anos.
As primeiras manifestações localizam-se na coluna vertebral e nas articulações proximais.
O quadro clínico caracteriza-se por sinais inflamatórios, por vezes acompanhados de expansões dolorosas, endurecimento dos tecidos periarticulares e perda progressiva da capacidade funcional da área afetada, sendo sua progressão no sentido axial-caudal e proximal-distal.
O quadro clínico caracteriza-se por sinais inflamatórios, por vezes acompanhados de expansões dolorosas, endurecimento dos tecidos periarticulares e perda progressiva da capacidade funcional da área afetada, sendo sua progressão no sentido axial-caudal e proximal-distal.
Destaca-se o início quase sempre na musculatura espinhal superior.
6. OSSO FOP x OSSO COMUM
A evolução da doença pode conduzir à fusão das articulações costocondrais e o envolvimento assimétrico da musculatura paravertebral pode determinar o aparecimento de escoliose e doença pulmonar restritiva, uma das causas de freqüente morbidade e mortalidade desta doença.
As radiografias convencionais documentam freqüentemente as anomalias esqueléticas constitucionais e as ossificações e anquiloses articulares correspondentes à história natural da doença.
O primeiro achado radiográfico é a presença de expansões ou massas em tecidos moles, que gradualmente diminuem de tamanho e ossificam.
A mineralização ocorre após três a quatro semanas, dando o aspecto final de colunas de osso que substituem os tecidos moles.
Pseudo-artroses também podem ocorrer com essas colunas de osso, principalmente nos ombros e no quadril.
A FOP deve ser prontamente identificada baseando-se apenas na história clínica, no exame físico e nos achados radiográficos.
Não deve ser realizado procedimento invasivo para determinação diagnóstica, pois tal fato é invariavelmente seguido de ossificações na região.
A deformidade mais característica desta doença e que sempre deverá suscitar a hipótese de FOP é o encurtamento bilateral com valgismo dos primeiros pododáctilos.
Más-formações da mão também podem estar associadas, sendo observado encurtamento do primeiro metacarpiano e braquimesofalangismo com clinodactilia do dedo mínimo.
7. HISTOPATOLOGIA – COMO FUNCIONAM OS OSSOS?
A histopatologia desta doença varia com o tempo de evolução das lesões e só existem alterações nas áreas anatômicas afetadas.
Este comportamento focal e evolutivo justifica a normalidade da biópsia obtida no paciente, numa fase inicial e provavelmente numa área não afetada.
Nas lesões precoces há, geralmente, infiltrado linfocitário, macrófagos e fibroblastos, evoluindo mais tarde para áreas de tecido conjuntivo com ossificação central, nas quais se distinguem osteoblastos, osteócitos e osteoclastos.
Recentemente, foi descrita a expressão aumentada da proteína morfogenética do osso 4 (BMP 4) nos fibroblastos presentes nas lesões precoces de FOP.
A proteína BMP 4 está localizada no cromossomo 14q22-q23, onde estão sendo pesquisadas evidências da existência de mutações neste gene ou na região de seu promotor.
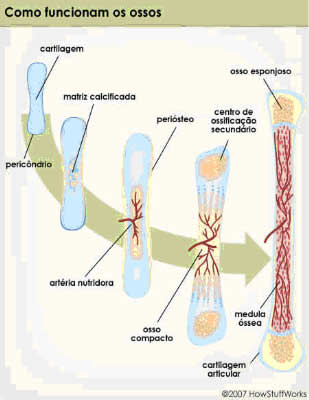 |
8. TRATAMENTO
Até o momento, não há tratamento conhecido e efetivo para esta doença. Toda conduta é conservadora e baseada no princípio do primum non nocere, evitando toda e qualquer condição potencialmente causadora de ossificação ectópica.
Existem diversos estudos envolvendo quelantes do cálcio, extratos de hormônio paratireóideo e vitamina D, que não parecem ser promissores.
Os corticóides poderão ser usados nos episódios de agudização inflamatória, embora não seja provada a inibição da ossificação ectópica. Etiodronato e isotretinoína também podem ser usados como inibidores da ossificação ectópica.
O diagnóstico diferencial desta entidade é bem limitado, uma vez que seu fenótipo, a história clínica e os achados radiográficos praticamente fecham o quadro.
Outras causas de ossificação ectópica devem sempre ser lembradas, tais como: osteodistrofia hereditária de Albright, calcificação heterotópica pseudomaligna, heteroplasia óssea progressiva e até mesmo o osteossarcoma.
A fisioterapia é caracterizada para este paciente por dar-lhe melhor qualidade de vida, no inicio com cinesioterapia, alongamentos e hidroterapia.
9. COMPLICAÇÕES
A FOP pode fazer com que ossos se formem ao redor do tórax, cercando os pulmões e dificultando a respiração. Essa condição é muito perigosa, e muitas vezes reduz o tempo de vida das pessoas com FOP.
10. ASSOCIAÇÃO INTERNACIONAL DE FOP - IFOPA.
IFOPA é uma rede de apoio a famílias que lidam com uma doença rara conhecida como FOP.
A Associação Internacional de Fibrodisplasia Ossificante Progressiva, ou IFOPA, é uma organização sem fins lucrativos que promove educação, comunicação e pesquisa médica.
Jeannie Peeper, uma mulher com FOP, criou a organização em 1988, porque queria reunir as pessoas com FOP. A partir deste início modesto, a IFOPA agora procura por pistas sobre a natureza desta doença em membros de mais de 40 países e apóia um respeitável laboratório de pesquisas na Universidade da Pensilvânia.
Graças à IFOPA e à pesquisa que ela patrocina, pessoas com FOP têm a esperança de que o futuro trará diagnósticos precoces, tratamentos eficientes e, eventualmente, a cura.
11. MUTTER MUSEUM: FILADÉLFIA - PENSILVÂNIA
O Museu foi fundado para educar futuros médicos sobre anatomia humana e anomalias médicas. Hoje, ela serve como um recurso valioso para educar o público e esclarecedor sobre o nosso passado e do médico dizendo importantes histórias sobre o que significa o ser humano.
Em exibição estão alguns objetos (20.000) apresentando anomalias terríveis para a saúde humana, incluindo um modelo de cera de uma mulher com um homem com um corno crescente na testa, um cinco-pé-longo cólon humano que continha mais de 40 quilos de matéria fecal, e do corpo petrificado a misteriosa Senhora Sabão, cujo cadáver foi transformado em uma substância chamada sabão adipocere.
O museu abriga também uma coleção de 2.000 objetos extraídos do povo da garganta, um tumor maligno removido do Presidente Grover Cleveland do palato duro, o fígado de conjoined siameses Chang and Eng Bunker e um crescimento removidas do tórax do assassino do presidente Abraham Lincoln, John Wilkes Booth.
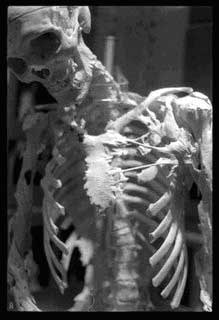
11.1. O esqueleto de Harry Eastlack - A mulher pedra
Harry Eastlack foi 'ossificada' ou 'pedra' pessoa. Ele sofria de uma doença rara conhecida como FOP (fibrodisplasia ossificante progressiva), que literalmente provém do tecido ósseo e imobiliza o paciente. Antes de morrer ele doou seu corpo para a ciência e seu único esqueleto está em exposição no Museu Mutter na Filadélfia.
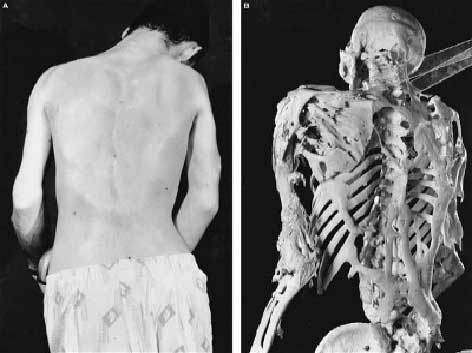 |
 |
 |






0 comentários:
Postar um comentário
Gostou? Reclame, elogie, sugira...